2.19 Kladen und komparative Daten
2.19.1 Hintergrund
In der letzten Einheit haben wir mit einer kurzen Einführung zu Stammbäumen in R begonnen. In dieser Einheit werden wir einige weitere Möglichkeiten erkunden, um Informationen aus einem Stammbaum zu extrahieren und sie zu visualisieren. Wir werden auch einen Blick darauf werfen, wie man Fragen zu vergleichenden Daten – Informationen zu Merkmalen oder andere Daten für einzelne Daten – beantworten kann, indem man sie auf einem Stammbaum darstellt.
2.19.2 Lernziele
Nach dieser Übungseinheit könnt ihr:
die Position der Wurzel eines Stammbaums verändern
bestimmte Kladen oder Spitzen auswählen oder hervorheben
vergleichende Daten mit einer Phylogenie verbinden und darstellen
eine Grafik mit geeignetem Format und Parametern speichern
2.19.3 Literatur
Liam Revell, Introduction to phylogenies in R
2.19.5 Tutorial
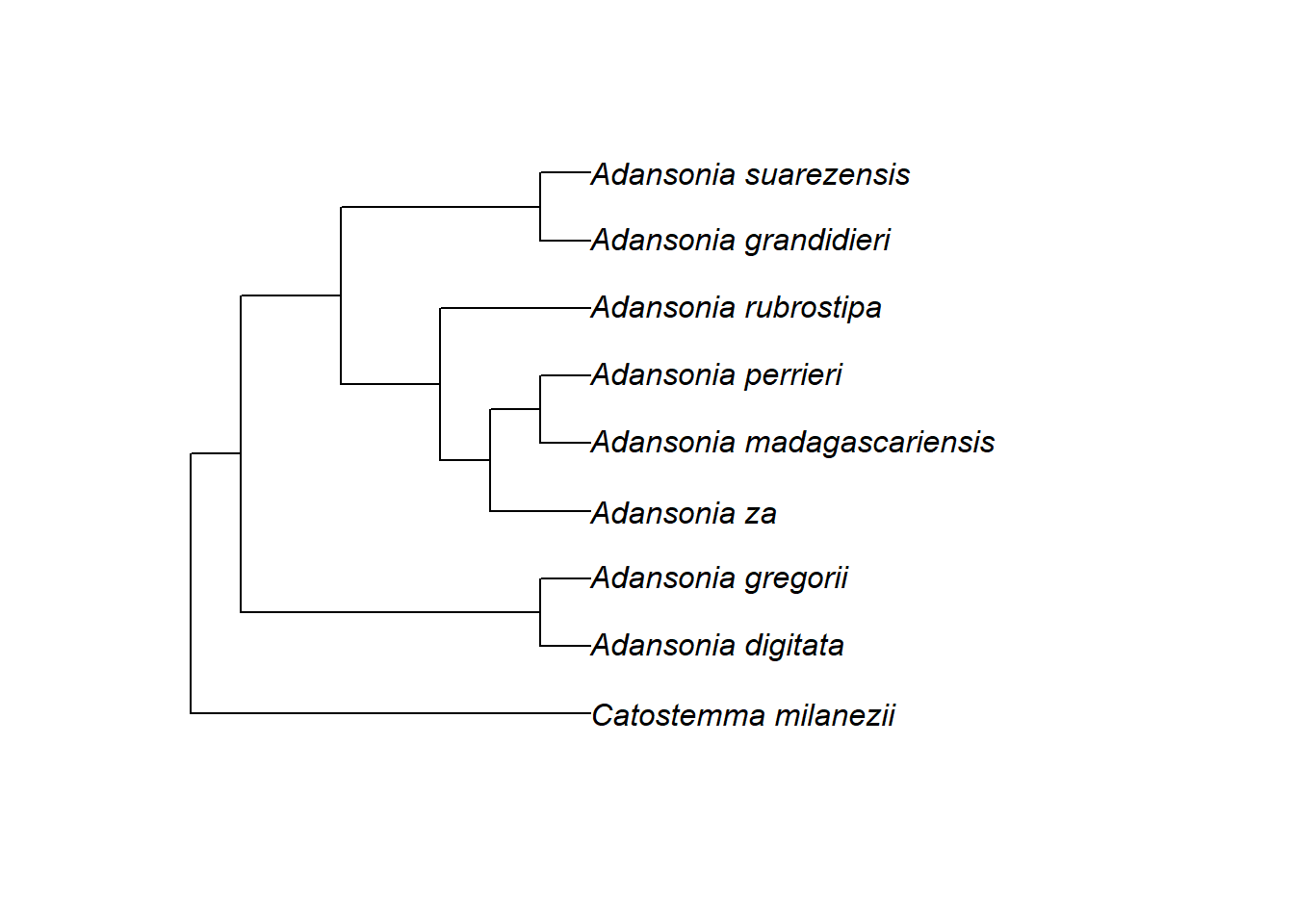
2.19.5.1 Bewurzelung und Anordnung der Äste
Wir haben in der letzten Einheit kurz gesehen, dass der älteste oder basalste Knoten in einem phylogenetischen Baum als Wurzelknoten oder einfach Wurzel bezeichnet wird. Dies ist die MRCA aller Spitzen in der Phylogenie. In einer Newick-Datei wird seine Position durch ein Semikolon angegeben.
Die Position der Wurzel kann man in der Regel nicht aus den Daten selbst ableiten, sondern muss sie aufgrund externer Informationen festlegen.
In diesem Fall war aus anderen Studien bekannt, dass die Arten von Adansonia enger miteinander verwandt sind als mit anderen Arten, so dass ein naher Verwandter – aus der Gattung Catostemma – als Außengruppe einbezogen wurde. Dadurch konnte die Wurzel an der Aufspaltung zwischen Adansonia und Catostemma platziert werden.
Wir könnten den Baum jedoch neu bewurzeln, wenn wir zum Beispiel der Meinung wären, dass Adansonia gregorii und A. digitata die außengruppe für alle anderen Spitzen sein sollten:
# die Außengruppe muss eine einzelne Spitze oder eine monophyletische Gruppe sein, sonst gibt R einen Fehler aus!
tree_rerooted <- root.phylo(phy = tree, outgroup = c("Adansonia_gregorii", "Adansonia_digitata"))
plot.phylo(tree_rerooted)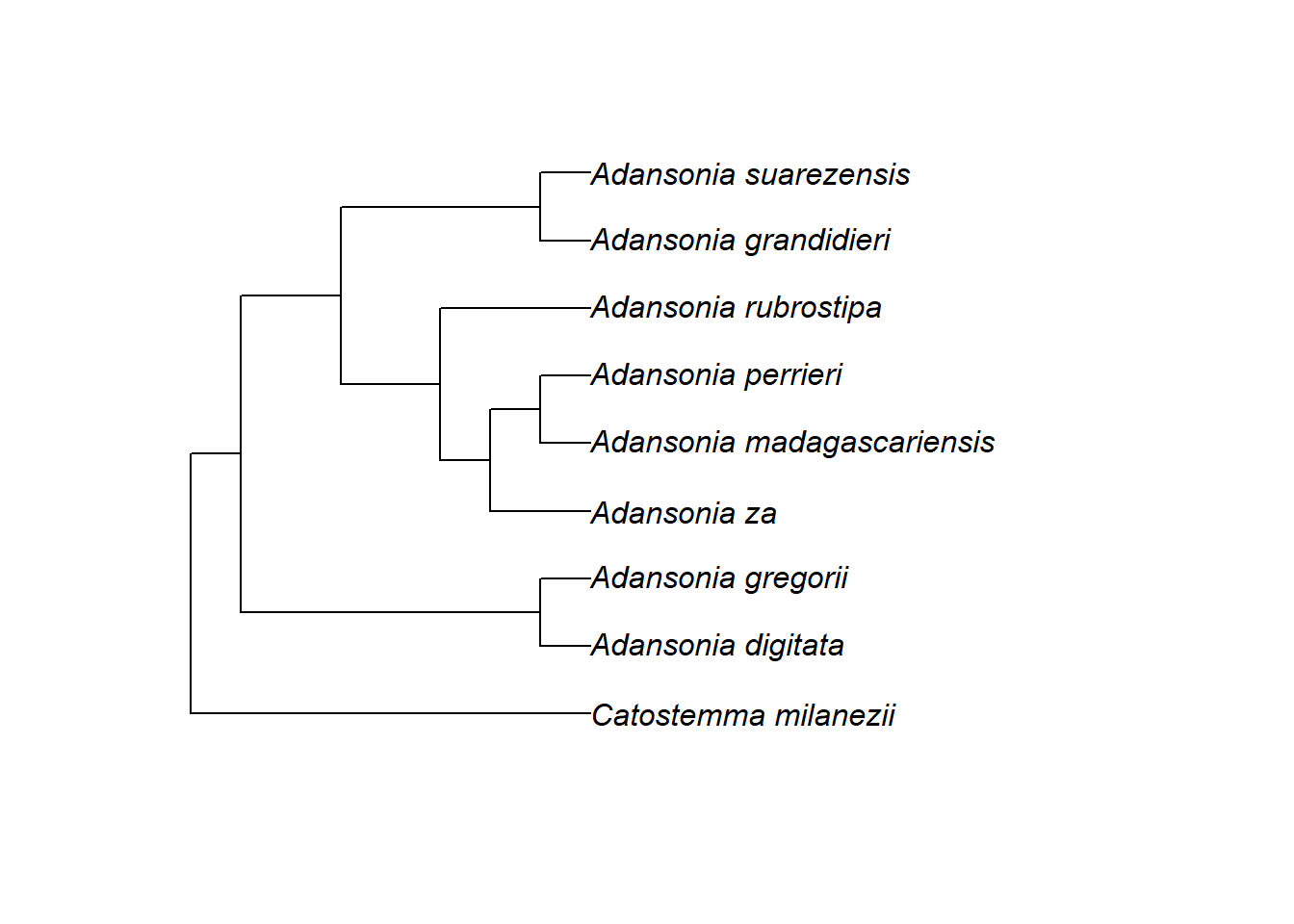
Dieser Stammbaum würde nun für eine ganz andere Evolutionsgeschichte stehen!
Wir können auch einfach die Zweige in der internen Struktur der Phylogenie neu anordnen, um eine ansprechendere Darstellung zu erhalten, indem wir die Funktion ladderize() verwenden. Dies ändert die Nummerierung der Knoten (man kann dies mit nodelabels() überprüfen), aber es repräsentiert immer noch die gleiche Evolutionsgeschichte (vergleiche den Stammbaum ganz oben)!
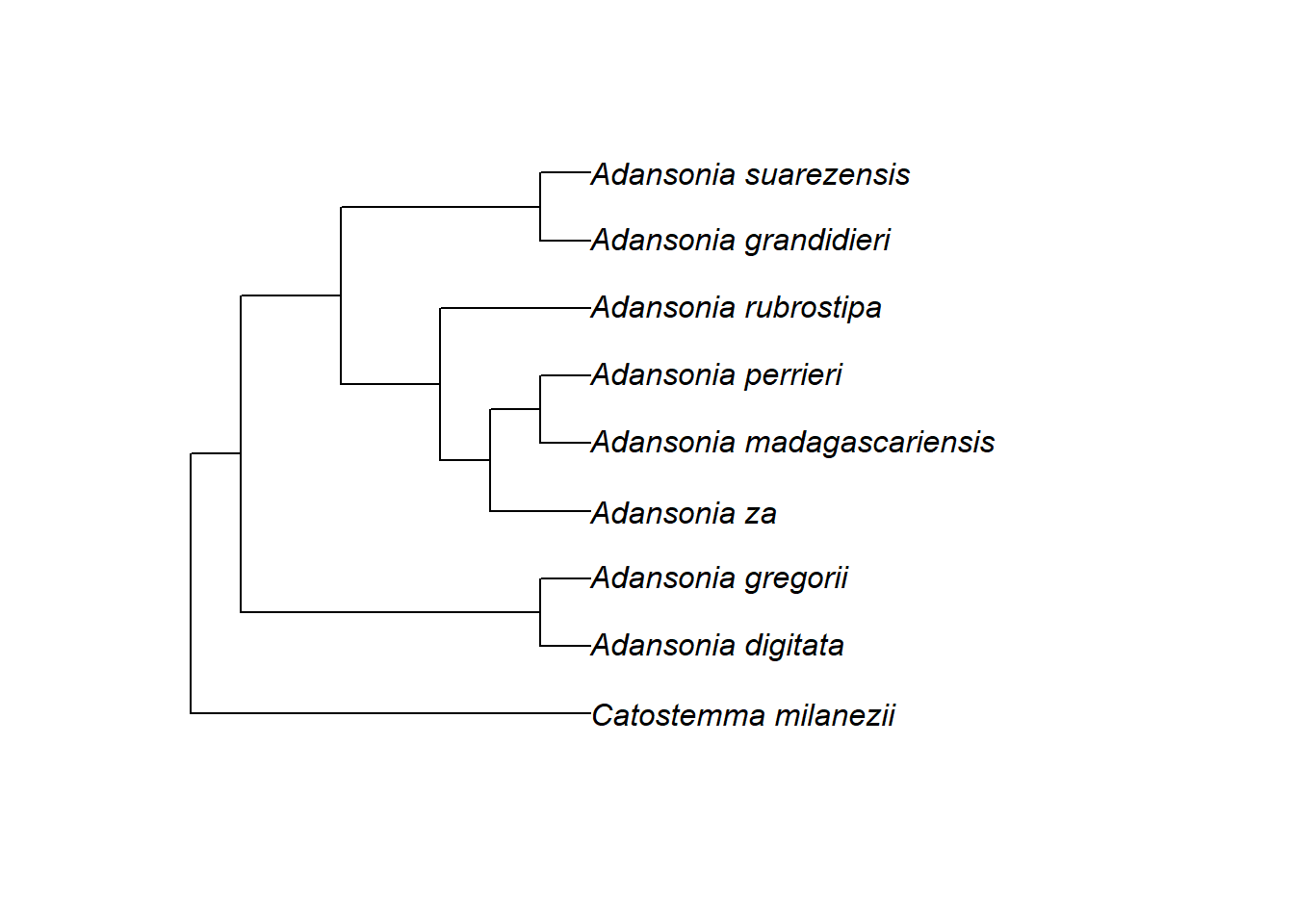
2.19.5.2 Spitzen hervorheben
Bisher haben wir uns einen Stammbaum der Gattung Adansonia angesehen. Heute werden wir einen größeren Baum verwenden, der auch Verwandte der Baobabs in der Unterfamilie Bombacoideae umfasst.
Die Datei bombacoideae.nwk enthält diesen größeren Stammbaum.
Ziemlich großer Stammbaum! Die Artnamen überlappen sich unter den Standardeinstellungen. Ihr könnt in RStudio in die Darstellung hineinzoomen, aber ihr könnt auch die Schriftgröße mit dem Argument cex (für “character expansion”) der Funktion plot.phylo() reduzieren, z.B. cex = 0.5.
Dieser Baum enthält Vertreter, aber nicht alle Arten, der Unterfamilie. Ihr seht, dass er auch einige nicht identifizierte Arten (“sp.”) und, als Außengruppe, zwei Arten von Sterculia, aus einer anderen Unterfamilie derselben Familie, enthält.
Nun möchten wir die Spitzen hervorheben, die zur Gattung Adansonia gehören. Wir können grep() verwenden, um alle Spitzen zu finden, die den Gattungsnamen enthalten, und dann erstellen wir einen Vektor von Schriftarten für die Darstellung der Spitzen:
g <- grep("Adansonia", tree$tip.label)
# grep() gibt uns die Positionen (Indizes) der Treffer in einem Zeichenvektor:
g## [1] 15 16 17 18 19 20 21 22# einen Vektor der Schriftarten für jede Spitze erstellen (3 bedeutet kursiv, die Standardeinstellung)
nt <- Ntip(tree)
font_type <- rep(3, length.out = nt)
# fett-kursive Schrift (4) für Adansonia-Spitzen verwenden
font_type[g] <- 4
# mit festgelegter Schriftart plotten
plot.phylo(tree, cex = 0.4, font = font_type)
2.19.5.3 Kladen auswählen
Nun möchten wir den Schwesterknoten der Baobabs (Adansonia) identifizieren.
Dazu benötigen wir eine Funktion aus einem anderen R-Paket, phytools. Dies ist ein sehr nützliches Paket für die Arbeit mit Stammbäumen, zusätzlich zu ape.
Benutzen wir also die Funktion getSisters() von phytools, um den Schwesterknoten von Adansonia zu finden:
library(phytools)
baobab_tips <- grep("Adansonia", tree$tip.label)
# Wir können einfach die Knotennummern der Spitzen (die Indizes) verwenden, um ihre MRCA zu erhalten:
baobab_mrca <- getMRCA(tree, tip = baobab_tips)
# den Schwesterknoten erhalten
baobab_sister <- getSisters(tree, node = baobab_mrca)
baobab_sister## [1] 137Wir möchten nun die Außengruppe, Sterculia, aus dem Stammbaum entfernen, um uns nur auf die eigentlichen Mitglieder der Bombacoideae-Klade zu konzentrieren. Wie können wir das tun?
2.19.6 Vergleichende Daten
Die Kombination eines Stammbaums mit Daten über Arten – zum Beispiel Merkmale oder geografische Vebreitung – ist der erste Schritt für viele Evolutions-Analysen.
Die Datei bombacoideae_distribution.csv enthält die Verbreitung der Bombacoideae-Arten in unserem Stammbaum. Es handelt sich um eine einfache Tabelle, die als Textdatei gespeichert ist, wobei die Spalten durch Kommas getrennt sind (CSV, comma-separated values).
Wahrscheinlich seid ihr bereits damit vertraut, wie man Daten aus einer CSV-Tabelle einliest:
# Ihr müsst das Spaltentrennzeichen angeben...
# ... und dass die erste Zeile die Spaltenüberschriften enthält.
tab <- read.table("./data/bombacoideae_distribution.csv", sep=",", header=TRUE)## [1] "data.frame"Wie alle Objekte, die in R geladen sind, erscheint tab oben rechts im RStudio-Fenster unter “Environment”. Ihr könnt darauf klicken, um es wie eine Excel-Tabelle anzuzeigen (dies entspricht dem Befehl View(tab)).
Die Spalte “tip” der Datentabelle ermöglicht es uns, die Daten den Baumspitzen zuzuordnen – wir können nicht einfach davon ausgehen, dass sie die gleichen Reihenfolge haben!
# match() findet die Positionen der ersten (exakten) Übereinstimmungen für die Werte eines Vektors in einem zweiten Vektor
m <- match(tree_ingroup$tip.label, tab$tip)
# Anhand dieser Positionen (Indizes) können wir die Zeilen der Tabelle neu anordnen, so dass sie dieselbe Reihenfolge haben wie die Spitzen des Stammbaums
tab <- tab[m,]Beachtet, wie wir [,] verwenden, um Zeilen oder Spalten einer Tabelle auszuwählen!
Jetzt, wo wir sie zugeordnet haben, können wir die Verbreitungswerte im Baum darstellen.
Wir erstellen einen Vektor mit Farben – schwarz für neotropische und weiß für paläotropische Arten:
# ifelse() erstellt einen Vektor abhängig davon, ob eine Bedingung erfüllt ist oder nicht
dis_colours = ifelse(tab$distribution == "neo", "black", "white")Mit diesem Vektor können wir dann die Verbreitung gegen die Spitzen auftragen:
# wir plotten den Baum zuerst, mit etwas Abstand zwischen den Spitzen und Ästen
plot.phylo(tree_ingroup, cex = 0.5, label.offset = 1)
# dann fügen wir Quadrate (pch=22) mit Hintergrundfarbe zu den Spitzen hinzu
tiplabels(pch = 22, bg = dis_colours)
Hinweis: Für die verschiedenen Symbole, die man in R plotten kann, z.B. mit tiplabels(), werft einen Blick auf die Hilfeseite von points().
Die Kombination solcher Vergleichsdaten mit dem Stammbaum kann uns helfen zu verstehen, wie sich ein Merkmal – eine Eigenschaft oder eine geografische Verteilung – entwickelt hat.
Ein Merkmal kann:
ursprünglich sein, wenn es bereits bei den Vorfahren einer bestimmten Gruppe vorhanden war, oder
abgeleitet, wenn es sich in einer bestimmten Gruppe neu entwickelt hat.
Die verwandten Begriffe Apomorphie, Plesiomorphie und Homoplasie haben wir bereits gesehen.
Natürlich können wir nur Hypothesen darüber aufstellen, was in der Vergangenheit passiert ist, da wir meist nur die heutige Verbreitung sehen (obwohl es möglich ist, fossile Taxa in einen Stammbaum mit aufzunehmen).
Zugegebenermaßen ist die Unterscheidung zwischen neotropischen und paläotropischen Arten nur eine grobe Darstellung ihrer geografischen Verbreitung. Idealerweise erstellt man genauere Verbreitungskarten, wie ihr es in den vorherigen Einheiten gesehen habt.
2.19.7 Grafiken speichern
Ihr könnt eine R-Grafik in RStudio speichern, indem ihr im Bereich Plots auf export klickt. Dies ermöglicht jedoch nur eine begrenzte Kontrolle über das Aussehen einer Abbildung.
Ihr könnt eine Abbildung in R mit Hilfe von Funktionen auch direkt in eine Datei schreiben. Auf diese Weise können ihr eure Befehle in einem Skript zusammenfassen und sie leicht wiederholen oder anpassen.
Für die meisten Abbildungen ist das Vektorformat SVG vorzuziehen, da es sich an jede Auflösung anpasst. Bei einer Abbildung mit vielen Elementen nimmt ein Rasterformat wie PNG jedoch oft weniger Platz ein. Ihr könnt Grafiken auch in ein PDF schreiben, aber das ist weniger praktisch, wenn man die Abbildung z.B. in ein Textdokument einbinden will.
Die entsprechenden Funktionen in R sind svg(), png() und pdf(). Sie öffnen ein “device”, das man dann nach der Ausführung der Plot-Funktionen wieder schließen muss:
# SVG öffnen, Format festlegen (standardmäßig in inches/Zoll)
svg(filename="../results/figure_bombacoideae.svg", width=8, height=12)
# par() legt grafische Parameter fest
# hier setzen wir den Rand auf 1/2 Textzeile auf allen vier Seiten
# – die Standardeinstellung erzeugt viel leeren Rand
par(mar = rep(0.5, 4))
# Stammbaum plotten (oder etwas anderes)
plot.phylo(tree_ingroup, cex=0.5, label.offset = 1)
tiplabels(pch = 21, bg = dis_colours)
# Grafik-device (hier SVG) schließen
dev.off()Wenn ihr die Grafik nun in einem Standard-Bildprogramm öffnet, könnt ihr die Auflösung in verschiedenen Zoom-Skalen überprüfen.
2.19.8 Aufgaben
- Stellt, unter Verwendung des obigen Beispiels und eurer Kenntnisse aus der vorherigen Einheit, den Stammbaum der Bombacoideae wie folgt dar: (1) die zur Gattung Pachira gehörenden Spitzen in blau (die anderen in schwarz) und (2) den MRCA-Knoten von Pachira mit einem blauen Punkt markiert.
Ist Pachira monophyletisch?
Was denkt ihr: Ist die paläotropische Verbreitung ursprünglich oder abgeleitet bei den Bombacoideae?
Reflektiert in 3-5 Sätzen, was ihr bisher über Stammbäume und deren Handhabung in R gelernt habt. Was kann man damit anfangen? Was ist schwierig zu verstehen?