2.22 Phylogenetische Inferenz und Datenbanken
Wir haben nun gesehen, wie man DNA als Daten nutzen kann, um die Verwandtschaftsbeziehungen zwischen Arten abzuleiten. Glücklicherweise gibt es inzwischen eine große Anzahl bereits verfügbarer Daten, die man nutzen kann. In dieser letzten Einheit sehen wir erstens, wie man DNA-Sequenzen aus Datenbanken zusammentragen kann. Zweitens werden wir sehen, wie man einen Stammbaum für eine Liste von Arten aus einem großen, verfügbaren Megastammbaum extrahieren kann.
2.22.1 Lernziele
Nach dieser Übungseinheit können Sie:
einen Satz von DNA-Sequenzen zur Analyse aus ENA auswählen und herunterladen
einen Stammbaum für eine Artenliste aus einem großen Megastammbaum extrahieren
2.22.3 Tutorial
2.22.3.1 DNA-Datenbanken
Die International Nucleotide Sequence Database Collaboration (INSDC) haben wir bereits gesehen. Sie besteht im wesentlichen aus drei DNA-Datenbanken (DDBJ aus Japan, GenBank aus den USA und das European Nucleotide Archive, ENA). Sie enthält die allermeisten Sequenzen, die jemals veröffentlicht wurden, da Wissenschaftler*innen neue Daten in der Regel an eine der teilnehmenden Datenbanken übermitteln müssen, wenn sie ihre Arbeit veröffentlichen.
Da die drei Datenbanken ihre Inhalte synchronisieren, kann man diese über jede der drei Webseiten abfragen. Im wesentlichen gibt es dafür zwei Herangehensweisen:
Wir haben eine DNA-Sequenz und möchten diese mit verwandten Sequenzen in einem Stammbaum platzieren. Dafür können die die Datenbank mit BLAST durchsuchen, wie wir es bereits gesehen haben.
Wir nutzen den Datenbank-Browser, um Daten für eine bestimmte Gruppe und ein bestimmtes Gen zu finden. Im ENA-Browser hat man die Option einer einfachen Suche, etwa “Bombacoideae matK”, oder man kann über Advanced Search diverse Filteroptionen nutzen.
2.22.3.2 Megastammbaum
Für diverse Organismengruppen gibt es bereits große “Mega-” oder “Rückgrat-”Stammbäume, die versuchen, alle zu diesem Zeitpunkt verfügbaren DNA-Sequenzen zu integrieren. Für die Samenpflanzen etwa haben Smith & Brown (2018) einen solchen Rückgrat-Stammbaum erstellt.
Dieser Stammbaum enthält nahezu alle zu diesem Zeitpunkt beschriebenen Arten und insgesamt 353.185 Spitzen. Davon sind aber nur etwa 70.000 tatsächlich auch Taxa, für die DNA-Sequenzen verfügbar waren. Die übrigen wurden mithilfe der Taxonomie von Open Tree of Life im Stammbaum platziert. Dadurch entstehen Polytomien, also Knoten, die nicht dichotom oder binär sind und damit mehr als zwei Tochterlinien haben.
Der Stammbaum ist als Datensatz mit dem Artikel verfügbar und hier als Newick-Datei SmithBrown_2019_ALLOTB.tre.
library(ape)
# Megastammbaum einlesen
megatree <- read.tree("./data/SmithBrown_2019_ALLOTB.tre")
# kurze Übersicht
megatree##
## Phylogenetic tree with 353185 tips and 85679 internal nodes.
##
## Tip labels:
## Humiriastrum_subcrenatum, Humiriastrum_dentatum, Humiriastrum_glaziovii, Humiriastrum_cuspidatum, Humiriastrum_diguense, Humiriastrum_excelsum, ...
## Node labels:
## Spermatophyta, Magnoliophyta, mrcaott2ott2645, mrcaott2ott35778, Mesangiospermae, mrcaott2ott121, ...
##
## Rooted; includes branch length(s).## [1] 353185## [1] 85679# ein komplett binärer/dichotomer Stammbaum hätte genau einen Knoten weniger als Spitzen
Ntip(megatree) - 1## [1] 353184## [1] FALSE# hier sind es deutlich weniger Knoten
# das liegt daran, dass der Baum nicht binär ist, also Polytomien enthält:
is.binary(megatree)## [1] FALSEDieser Mega-Stammbaum ist leider so groß, dass man ihn nicht sinnvoll grafisch anzeigen kann – R wäre vermutlich überfordert und man würde den Baum vor lauter Ästen nicht sehen! Daher müssen wir durch Funktionen einzelne Bereiche auswählen.
2.22.3.3 Spitzen exakt auswählen
Wir können zum Beispiel mit keep.tip() einen Teil des Stammbaums, der eine Liste exakter Artnamen verbindet, auswählen. Als Beispiel der Stammbaum der Nutzpflanzen, die man in einer typischen Pizza finden könnte:
# Vektor mit Artnamen definieren
sp <- c("Triticum_aestivum", "Olea_europaea", "Origanum_vulgare",
"Allium_tuberosum", "Allium_cepa_var._aggregatum", "Capsicum_annuum",
"Solanum_lycopersicum", "Solanum_melongena",
"Eruca_vesicaria_subsp._sativa")
# neuen Stammbaum definieren, der nur diese Arten aus dem Megastammbaum enthält
tree <- keep.tip(megatree, tip = sp)
# Stammbaum anzeigen
plot.phylo(tree)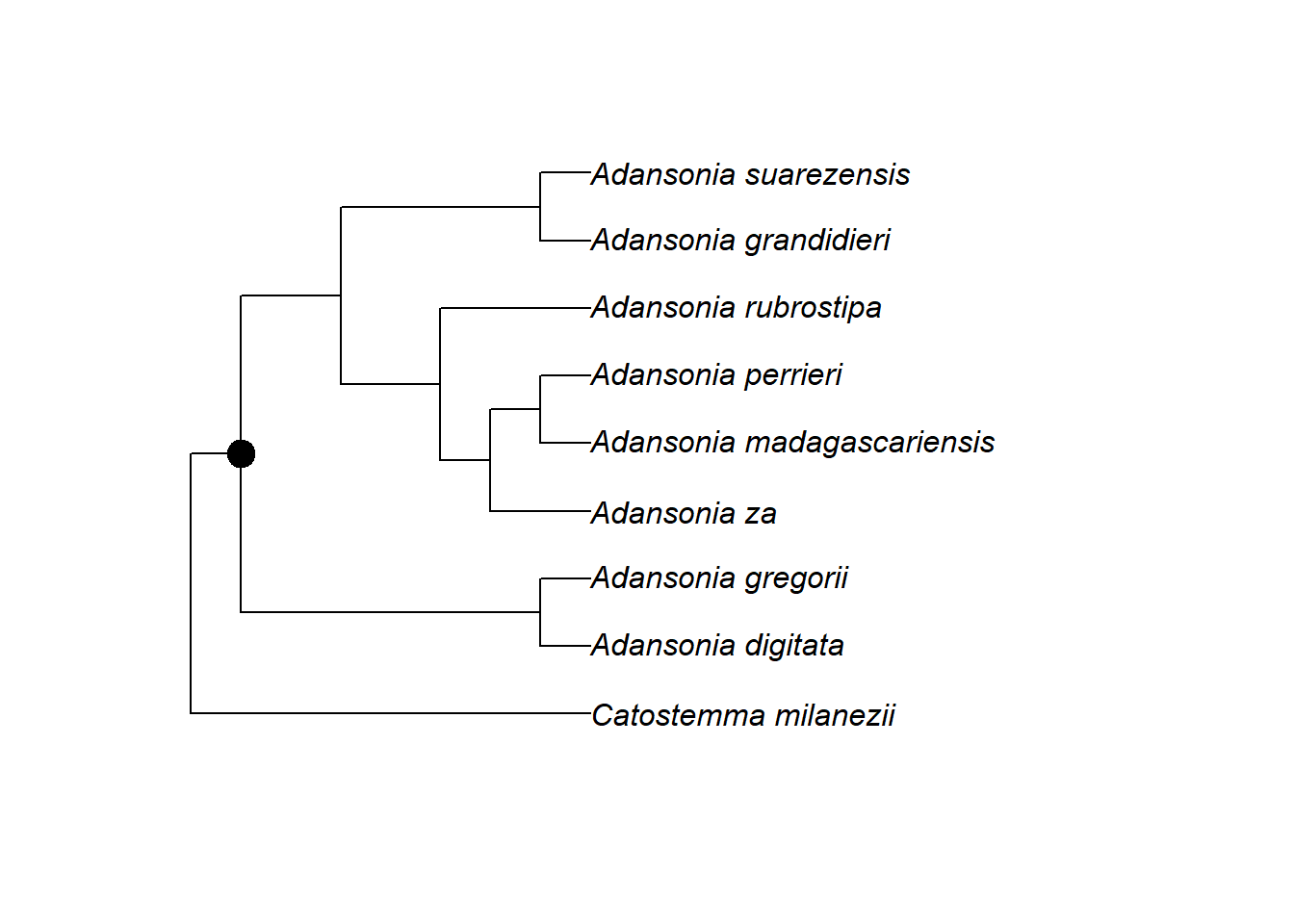
Wichtig: Diese Vorgehensweise funktioniert nur mit Namen, die exakt so im Stammbaum enthalten sind (inklusive der Unterstriche). Manchmal sind Unterarten oder Varietäten enthalten; hier hätte z.B. “Eruca_vesicaria” nicht funktioniert. Auch muss man, wenn man eine gewünschte Art nicht findet, eventuell über Plants of the World Online überprüfen, ob im Megastammbaum vielleicht ein veraltetes Synonym verwendet wird.
Einen exakten Treffer findet man mit match(), während grep() Treffer findet, die ein Muster enthalten. Damit kann man Arten schrittweise finden:
## [1] NA## [1] 9## [1] "Allium_cepa_var._aggregatum"## [1] "Adansonia_gregorii" "Adansonia_kilima"
## [3] "Adansonia_digitata" "Adansonia_za"
## [5] "Adansonia_perrieri" "Adansonia_madagascariensis"
## [7] "Adansonia_rubrostipa" "Adansonia_suarezensis"
## [9] "Adansonia_grandidieri"Praktischer ist es, Artnamen aus einer Liste einzulesen, die man zuvor als Textdatei (etwa mit Editor) erstellt hat. In dieser Beispiel-Datei sind es Arten, die man bei uns auf einer typischen Fettwiese finden könnte:
# Artliste einlesen - jede Zeile wird als ein Element eines Vektors geladen
sp <- scan("./data/fettwiese_arten.txt", what = "character")
# Vektor anzeigen
sp## [1] "Arrhenatherum_elatius" "Dactylis_glomerata"
## [3] "Festuca_pratensis_subsp._pratensis" "Galium_mollugo"
## [5] "Heracleum_sphondylium" "Poa_pratensis_subsp._pratensis"
## [7] "Rumex_crispus" "Taraxacum_officinale"
## [9] "Trifolium_pratense" "Trifolium_repens"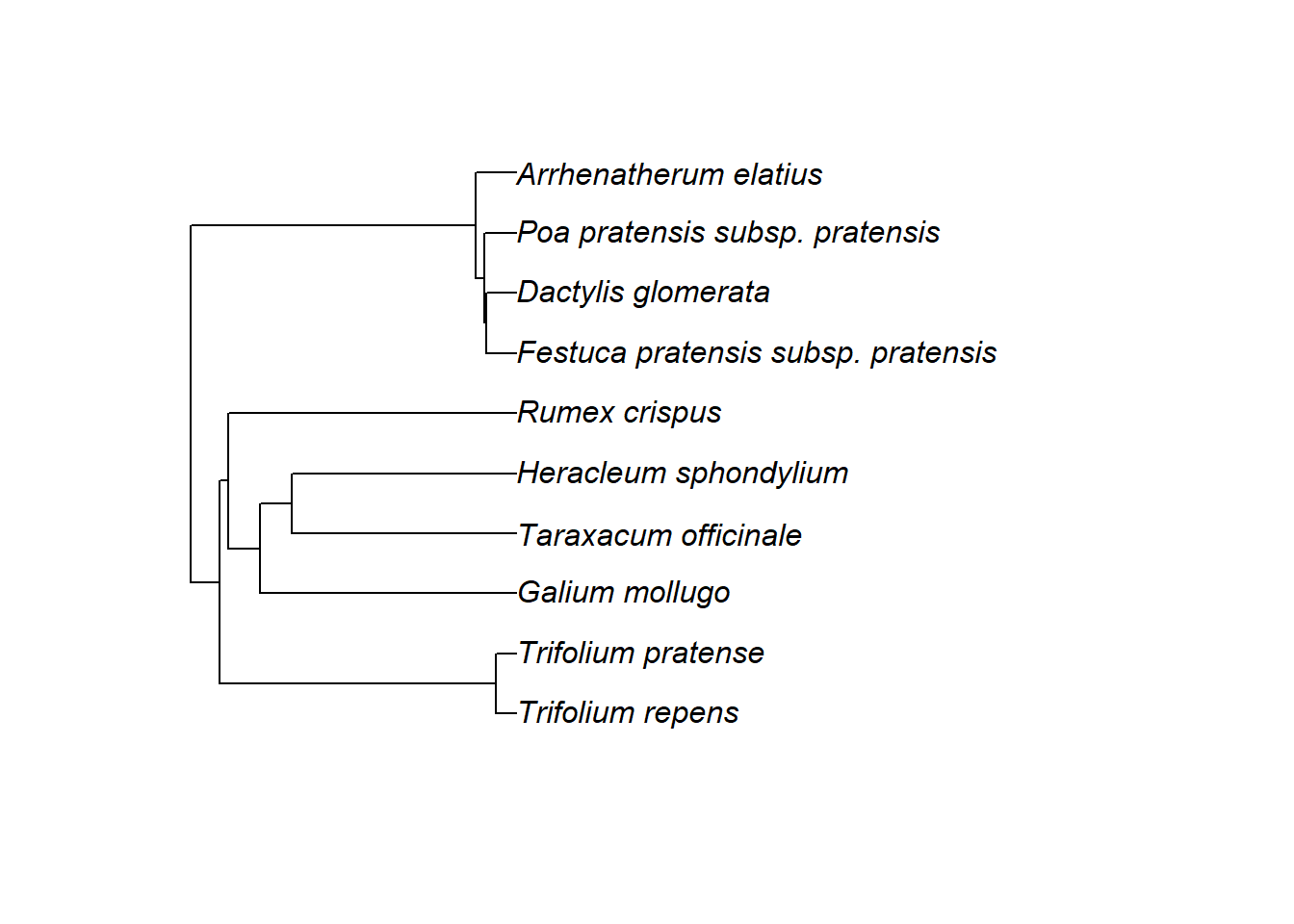
2.22.3.4 Spitzen nach Muster auswählen
Wir können mit grep() natürlich auch z.B. eine oder mehrere Gattungen auswählen. Hier etwa Echinacea und Zinnia, die beide innerhalb der Korbblütler nah verwandt sind:
# Muster suchen
g <- grep("Echinacea|Zinnia", megatree$tip.label)
# Stammbaum extrahieren
tree <- keep.tip(megatree, tip = g)
# Stammbaum anzeigen
plot.phylo(tree)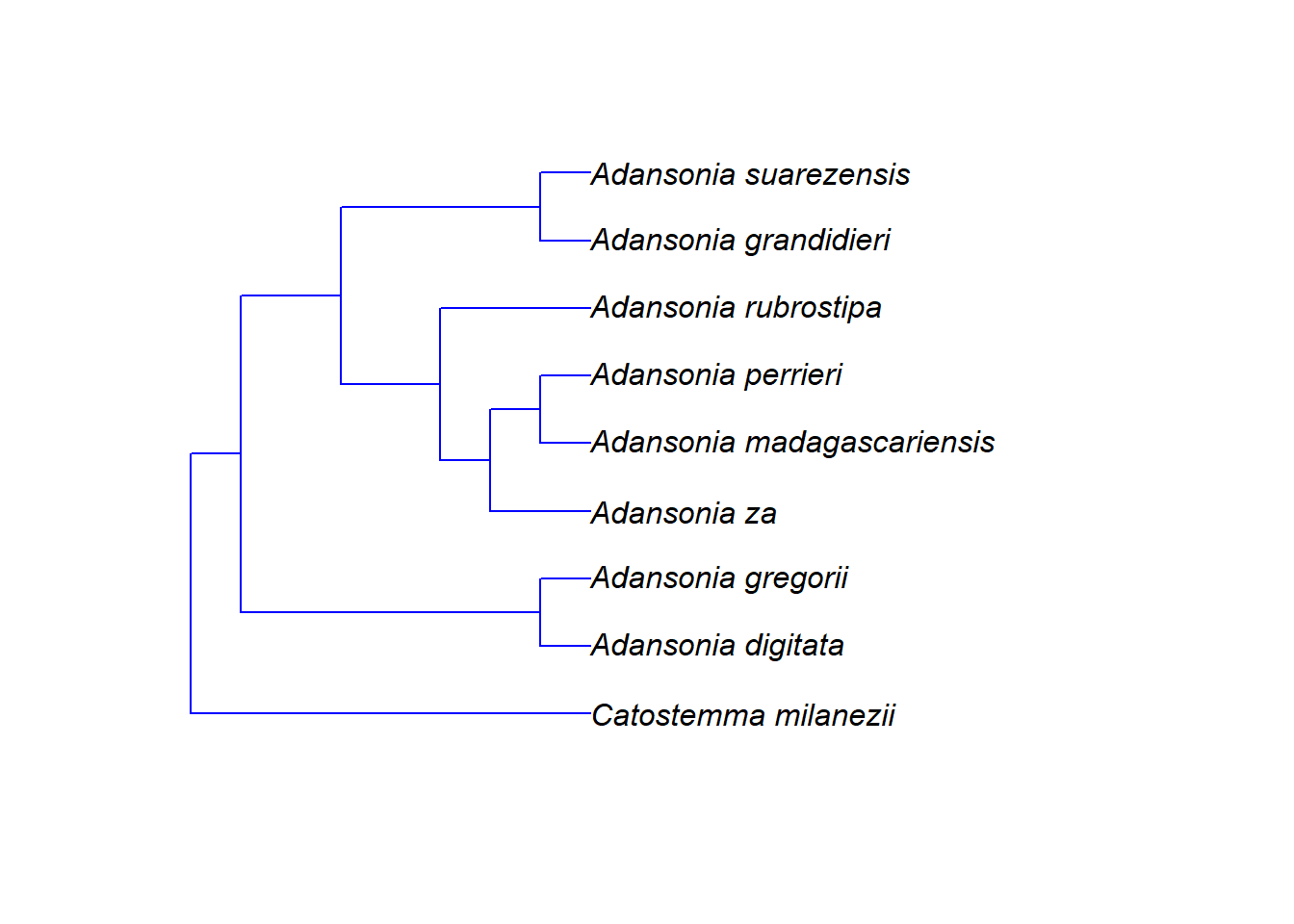
In diesem Beispiel sehen wir, dass die beiden Gattungen jeweils eine Polytomie bilden. In diesem Fall bedeutet das, dass keine DNA-Daten für diese Arten verfügbar waren, sondern sie einfach per Taxonomie dem Knoten ihrer Gattung zugewiesen wurden.
2.22.3.5 Knoten auswählen
Man kann außerdem einen Stammbaum per Knoten mit extract.clade() extrahieren. Da wir bei diesem Megastammbaum nicht einfach die Knotennummern grafisch anzeigen können (das geht bei einem kleinen Stammbaum mit nodelabels()), müssen wir den gewünschten Knoten durch Funktionen finden:
# Muster in den "tip labels" finden
g <- grep("Adansonia|Catostemma", megatree$tip.label)
# Knoten des gemeinsamen Vorfahrens der gefundenen Arten finden
n <- getMRCA(megatree, tip = g)
# Stammbaum über Knotennummer extrahieren
tree <- extract.clade(megatree, node = n)
# Stammbaum anzeigen
plot.phylo(tree)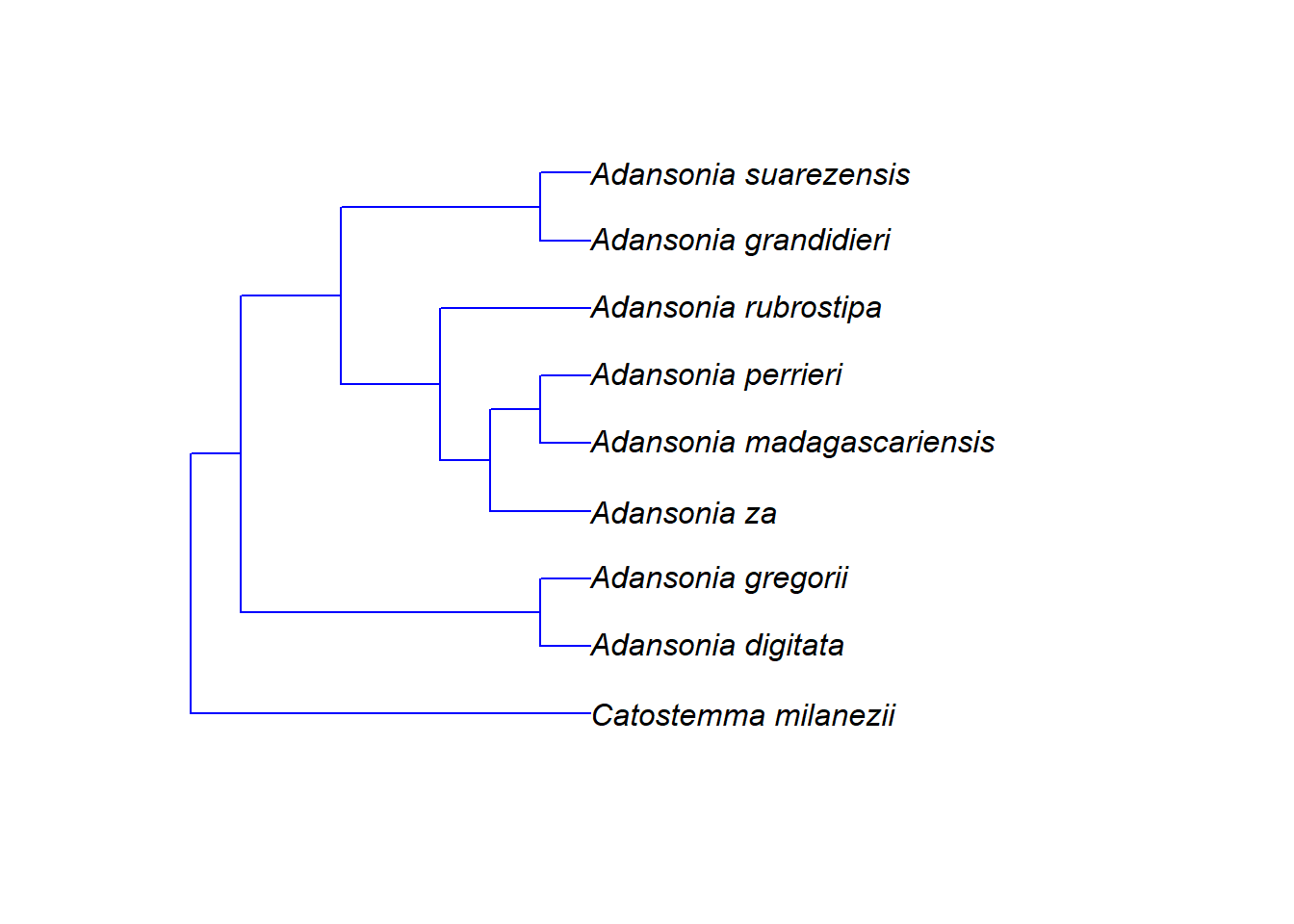
2.22.3.6 Geologisches Alter anzeigen
Der Megastammbaum ist außerdem geologisch datiert, das heißt, dass die Astlängen dem geschätzten Alter (in Millionen Jahren) entsprechen. Wir können daher z.B. für den oben extrahierten Stammbaum von Adansonia und seinen nächsten Verwandten eine Zeitskala hinzufügen:
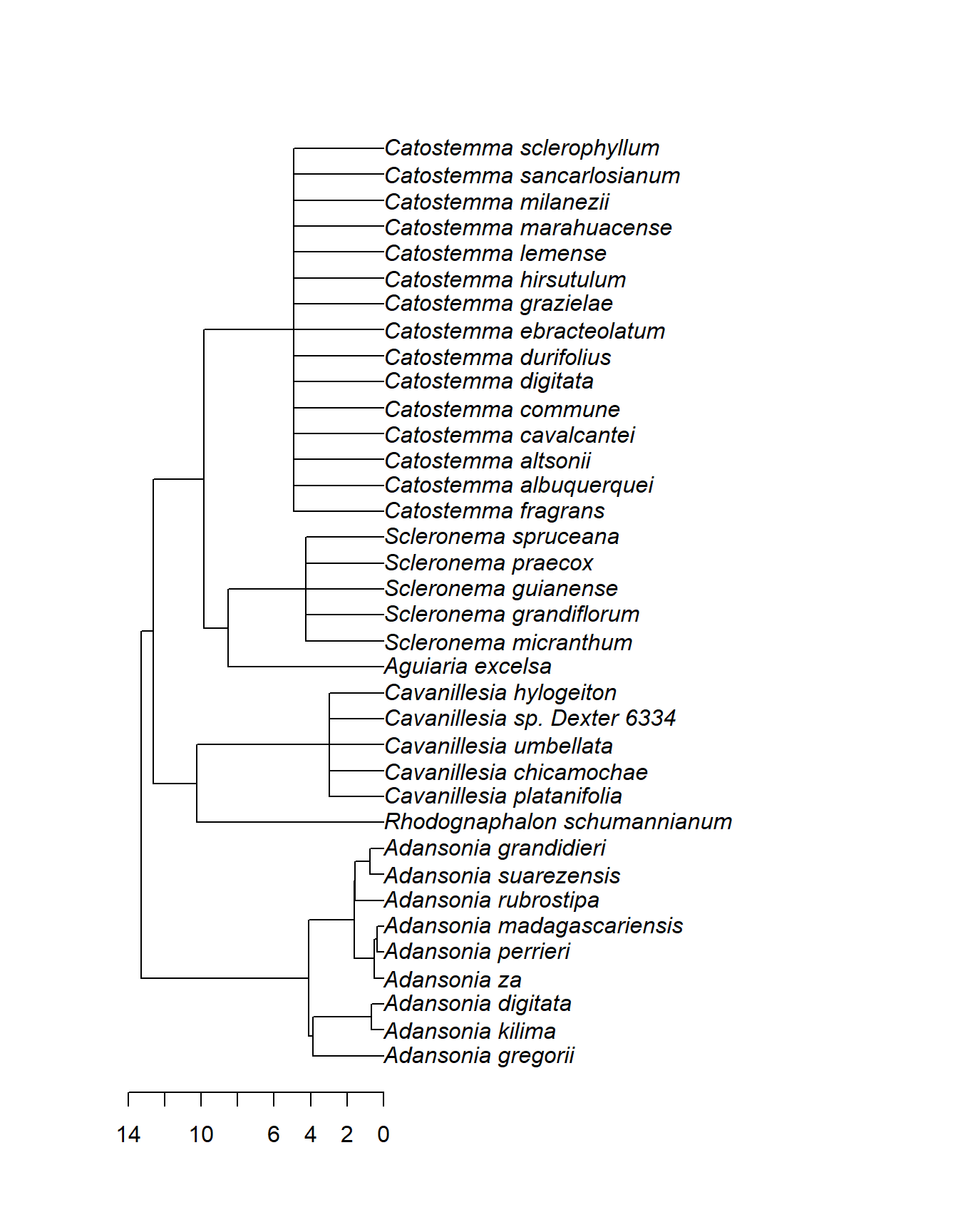
Nach dieser Skala hat sich die Gattung Adansonia vor etwa 14 Mio. Jahren von ihrer Schwestergruppe getrennt. Die Arten der Gattung selbst haben vor etwa 4 Mio. Jahren begonnen, sich aufzuspalten.
Aber Vorsicht! Bei Polytomien macht das Alter der Knoten nur begrenzt Sinn. Im Beispiel oben ist etwa die Gattung Catostemma ungefähr 5 Mio. Jahre alt. Wie wissen aber nicht, wann sich deren Arten untereinander später aufgetrennt haben – sicher nicht alle zum selben Zeitpunkt. Aber da keine DNA-Daten verfügbar waren, können wir keine genauere Aussage darüber treffen.
Außerdem entspricht die Zeitachse des Megastammbaums nur groben Schätzwerten. In anderen Studien könnten sich deutliche Abweichungen ergeben.
2.22.4 Aufgaben
Wählen Sie eine Pflanzengattung und eine nah verwandte Außengruppe aus (das erfordert etwas Internetrecherche). Suchen Sie in der ENA-Datenbank nach Sequenzen von rbcL (ein häufig verwendetes Chloroplasten-Gen) für 4-6 Arten der Gattung und 1-2 Arten der Außengruppe. Laden Sie die Sequenzen herunter und erstellen Sie einen Stammbaum mit den Zwischenschritten, die Sie in den vorherigen Einheiten gesehen haben.
Erstellen Sie eine Liste von 5-8 Pflanzenarten, die Sie selbst im Gelände gesehen haben. Extrahieren Sie den Stammbaum dieser Arten aus der Smith-&-Brown-Megaphylogenie. Stellen Sie diesen Stammbaum grafisch dar, aber ersetzen Sie die wissenschaftlichen Namen durch die Trivialnamen (dazu können Sie Wissen aus der Einheit Stammbäume in R nutzen).
Wie können Sie feststellen, wann sich die Gattungen Oryza (Reis) und Zea (Mais) voneinander getrennt haben?