2.20 DNA und Alignment
2.20.1 Hintergrund
Sie haben nun die Grundlagen des Umgangs mit phylogenetischen Daten in R kennengelernt. Aber wie leiten wir eigentlich einen Stammbaum für eine Gruppe von Arten aus Daten ab? In dieser Einheit geht es um den ersten grundlegenden Schritt in der phylogenetischen Inferenz: die Inferenz von Homologie. Wir werden dies für DNA-Sequenzen mittels DNA-Alignment tun. Wir werden R verwenden, aber auch MEGA, eine freie, grafische Software für Analysen von DNA und Stammbäumen.
2.20.2 Lernziele
Nach dieser Übungseinheit werden Sie in der Lage sein:
den Begriff Homologie im Zusammenhang mit DNA-Basen zu erklären
Indels und Substitutionen in einem DNA-Sequenz-Alignment zu identifizieren
Paarweises und multiples Alignment auf einen Satz von DNA-Sequenzen anzuwenden
die genetischen Differenzen zwischen DNA-Sequenzen zu interpretieren
2.20.3 Literatur/Weiterführendes
What is sequence alignment (12 min) by Kevin Yip, Chinese University of Hong Kong.
2.20.5 Tutorial
2.20.5.1 Homologie
Wenn wir wissen wollen, wie eine Gruppe von Arten (oder Genen, Viren, Proteinen etc.) miteinander verwandt ist, brauchen wir etwas, womit wir sie vergleichen können. Dieses “Etwas” muss unterschiedlich genug sein, um uns zu erlauben, Gruppen zu bilden, aber ähnlich genug, um sagen zu können, dass wir etwas der gleichen Sorte vergleichen.
Wir suchen nach Dingen, die den gleichen Ursprung haben, was bedeutet, dass sie homolog sind. Das bedeutet nicht, dass sie sehr ähnlich sein müssen; sie können im Laufe der Zeit durch die Evolution stark verändert werden. Im Gegensatz dazu sind zwei Dinge analog, wenn sie ein ähnliches Aussehen oder eine ähnliche Funktion haben, diesen Zustand aber durch unabhängige evolutionäre Ursprünge erreicht haben.
_Nepenthes_Veitchii_Pink_(49360096312)__geoff_mckay_CC_BY-2.0.jpg) Nepenthes-Kannenfallen Nepenthes-Kannenfallen |
 Sarracenia-Schlauchfallen Sarracenia-Schlauchfallen |
Homologie ist immer auf einer bestimmten Hierarchieebene angesiedelt. Bei den Pflanzen haben sich beispielsweise viele verschiedene Blattmodifikationen entwickelt, wie z. B. Kannen bei einigen fleischfressenden Pflanzen oder Dornen bei Kakteen. Auf der Ebene des Blattes sind sie homolog, da sie alle von derselben ursprünglichen Struktur abgeleitet sind.
Die fleischfressenden Pflanzengattungen Sarracenia und Nepenthes sind nur entfernt verwandt, und ihre Fallgruben-Fallen (Kannen oder Schläuche) haben sich unabhängig voneinander entwickelt. Auf der Ebene der Falle sind diese Blattveränderungen daher analog, da sie nicht auf einen gemeinsamen Vorfahren zurückgeführt werden können.
2.20.5.2 DNA-Sequenzen als Daten
Für phylogenetische Zwecke ist DNA der am häufigsten verwendete Datentyp. Sie hat Millionen von Positionen (sites), die wir potenziell vergleichen können, aber einen festen Satz von vier Zeichen (characters), die DNA-Basen oder Nukleotide, die wir leicht als Text darstellen können: A, T, G und C.
Normalerweise vergleichen wir DNA-Sequenzen, die einen gemeinsamen Ursprung haben, z. B. ein Gen, das von einem Vorfahren einer Gruppe an alle seine Nachkommen weitergegeben wurde. Dieser spezielle Fall von Homologie wird bei DNA-Sequenzen Orthologie genannt. (Schwierig wird es, wenn sich die Geschichte der Gene von der Geschichte der Art durch Paralogie unterscheidet, aber das klammern wir hier erst einmal aus.)
Die einzelnen Stellen in DNA-Sequenzen entwickeln sich durch Mutationen, was die Feststellung der Homologie innerhalb der Sequenz erschweren kann. Mutationen können Substitutionen sein, bei denen Zeichen an einer Stelle ersetzt werden, aber auch Insertionen und Deletionen, bei denen Stellen hinzugefügt werden oder verschwinden. Eine Veränderung durch Insertion oder Deletion wird als Indel bezeichnet: Oft lässt sich nicht feststellen, welche der beiden Prozessen die Veränderung verursacht hat. Indels können die Länge von DNA-Sequenzen verändern.
2.20.5.3 Das FASTA-Format
Wie für Stammbäume gibt es auch für DNA-Sequenzen standardisierte Textformate. Sie können sowohl für die eigentlichen Sequenzen (die nicht alle die gleiche Länge haben) als auch für durch Alignment ausgerichtete Sequenzen (mit Lücken und alle gleich lang) verwendet werden.
Das gebräuchlichste Format ist FASTA:
>[Sequenz_1_Name] [Beschreibung]
CGGCTTAGGCCTTTGAC
>[Sequenz_1_Name] [Beschreibung]
ACGGTAGGTCTTCGTAC
...Der Sequenzname darf keine Leerzeichen enthalten. Für die DNA-Sequenzen sind sowohl Großbuchstaben (ATCG) als auch Kleinbuchstaben (atcg) zulässig. Mehrdeutige Zeichen werden nach einem Mehrdeutigkeitscode kodiert: N bedeutet beispielsweise eine der vier DNA-Basen. Die Sequenzen können in mehrere Zeilen aufgeteilt und durch Leerzeichen unterbrochen werden, z. B. um sie in Spalten anzuordnen.
2.20.5.4 DNA-Alignment
Um homologe DNA-Zeichen vergleichen zu können, müssen wir die DNA-Sequenzen durch Alignment ausrichten. Das bedeutet, dass wir Lücken (gaps) für alle Indels einführen, die wir ableiten (oder fehlende Daten an den linken oder rechten Enden):
-CGGCTTAGGCCTTTG-AC
ACGG--TAGGTCTTCGTACWir sehen Folgendes:
In einem Alignment haben alle Sequenzen die gleiche Länge. Stellen Sie sich das Ganze wie eine Datenmatrix oder Tabelle vor, mit Proben (oft eine pro Art) als Zeilen und DNA-Positionen als Spalten.
Wir können auf eine Mindestzahl von Mutationen schließen, aber natürlich können zwei Zeichen durch viele unabhängige, versteckte Veränderungen, die zum gleichen Ergebnis führten, identisch sein (Homoplasie). Das bedeutet auch, dass wir nie ganz sicher sein können, dass die Zeichen in einer Alignment-Spalte homolog sind – wir können nur eine vernünftige Annäherung finden.
Viele Algorithmen versuchen, ein Alignment zu finden, das die Anzahl der Änderungen, die man annehmen muss, minimiert. Dies ist ein allgemeiner Grundsatz, Parsimonie, der in der Phylogenetik häufig anzutreffen ist.
Die meisten Algorithmen gehen davon aus, dass Indels unwahrscheinlicher sind als Substitutionen, und weisen ihnen daher höhere Kosten bzw. einen höheren Malus zu.
2.20.5.5 Paarweises Alignment: BLAST
Das basic local alignment search tool (BLAST) ist ein äußerst nützlicher und schneller Algorithmus für den Abgleich zweier Sequenzen. Er wird in der Regel verwendet, um eine Abfrage-Sequenz mit einer großen Referenz-Sequenz (z. B. einem vollständigen Genom) oder einer Referenzdatenbank zu vergleichen und die ähnlichste Sequenz zu finden.
Die International Nucleotide Sequence Database Collaboration (INSDC) ist eine weltweite Sammlung von DNA-Sequenzen. Sie enthält die allermeisten Sequenzen, die jemals veröffentlicht wurden, da Wissenschaftler*innen neue Daten in der Regel an eine der teilnehmenden Datenbanken übermitteln müssen, wenn sie ihre Arbeit veröffentlichen.
Wir können den BLAST-Algorithmus online verwenden, um eine Abfragesequenz mit den Daten in INSDC zu vergleichen. Die FASTA-Datei matk_unknown_bombacoideae.fas enthält einen kurzen Abschnitt des Maturase K (matK)-Chloroplastengens, das für eine unbekannte, im Feld gesammelte Bombacoideae-Art sequenziert wurde:
## >Bombacoideae_unknown_sp.
## TTTTTTTATGAGTCCTAATTATTTATTCCCTTTATGGGTTAGACATGAATGTGTAGAAAAAGCAGTATATTGATAAAGAAAAGAAATTTTGTTTCCAAAATCAAAAGAGCGATTGGGTTGAAAAAATAAAGGATTTCTAACCATCTTCTTATCCTATAACGAACATAAATCAATTAGATGGCAAAAGATAGGATAGAGAATCCGTTGATGAATCTACCTGTCTCCGAGGATCTATTCTTTTCTTACTATAATACCTTGTTTTGACTGTATCGCACTATGTATCATTTGATAACCGAATAGATCCCCTATCTTTGGTTCAAATCGAATTTGAAATGGAGGAATTTCAAGTATATTTAGAACTAAATAGATCTCGTCGACATGATTTCCTATACCCACTTATTTTTCGGGAGTATATTTATGCACTTGCTCATGATCATGGTTTAAATAAATCGATGATTTTTTTGGAAAATCAGGGTTATGTTTATAAATTCAGTTCACTAATTGTGAAACGTTTAATTATTCGAATGGATCAACAGAATCATTTAATTATTTCTGCTAATGATTCCAACCAAAATCCATTTTTTGGGCACAACAATAATTTGTATTCTCAAATGATATCGGCGGGATTTGCAGTCATTGTGGAAATTCCATTTTCCTTACGATTAGTATCTTACTCACAAGGGGAAGAAGTCGCAAAATCTCATAATTTACAATCAATTCATTCAATATTTCCTTTTTTAGAGGACAAATTCTCACATTTAAATTATGTGTTAGATGTACTAATACCTCACCCCATCCATCTAGAAATCTTGGTTCAAGCCCTTCGCTACTGGGTAAAAGATGCTTCTTCTTTGCATTTATTACGGTTCTCTCTCTACWir können sie nun mit allen in INSDC hinterlegten Bombacoideae-Sequenzen vergleichen. Dafür gehen wir zur BLAST-Seite von GenBank, dem US-amerikanischen Teil von INSDC. Unter “blastn” laden wir die unbekannte Sequenz hoch oder fügen Sie sie ein. Weil wir schon wissen, dass die Sequenz zur Unterfamilie Bombacoideae gehört, beschränken wir die Suche unter “Organism” entsprechend. Wir belassen die Standardeinstellungen für alle anderen Parameter und klicken dann auf “BLAST”:
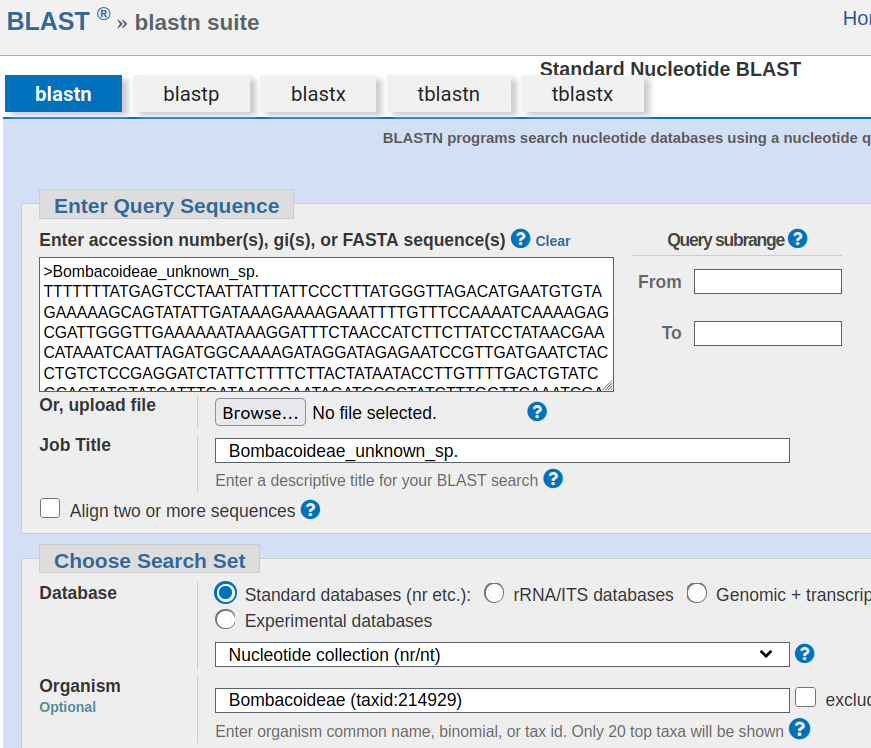
Der blastn-Algorithmus vergleicht zwei Nukleotid-/DNA-Sequenzen, aber wir könnten auch Proteinsequenzen vergleichen. Das Laden der Ergebnisseite kann je nach Benutzeraufkommen einige Sekunden oder Minuten dauern.
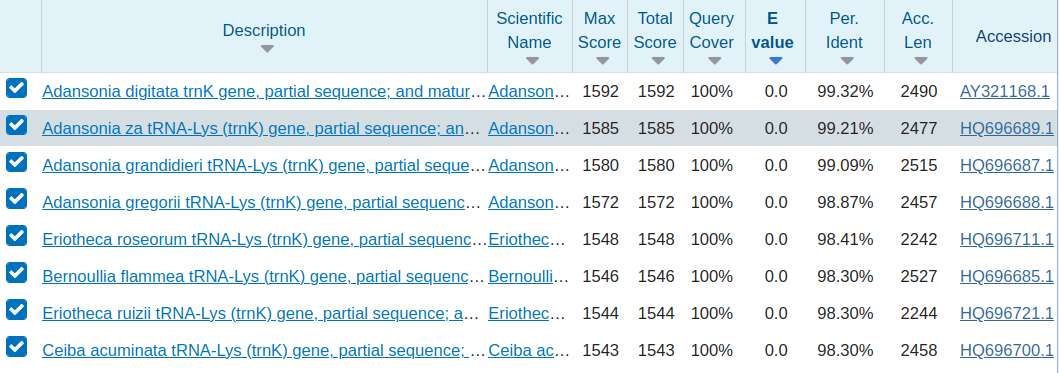
Die Ergebnistabelle zeigt die besten Übereinstimmungen, sortiert nach prozentualer Ähnlichkeit. Man sieht auch, dass BLAST für jedes paarweise Alignment einen Score und einen “e-Wert” berechnet; dieser entspricht wie warscheinlich es ist, einen Treffer mit ähnlicher Qualität durch reinen Zufall zu finden. Wir sehen auch auch, ob die gesamte oder nur ein Teil der Suchsequenz abgeglichen werden konnte (Query Cover).
Wenn Sie auf das beste match klicken, erhalten Sie eine Ansicht des paarweisen Alignments.
2.20.5.6 Multiples Sequenzalignment in MEGA
Das Alignment mehrerer Sequenzen ist eine viel komplexere Aufgabe als das Alignment von zwei Sequenzen. Es erfordert in der Regel Algorithmen mit mehreren Verbesserungsrunden.
Hinweis: Multiples Sequenzalignment ist im Allgemeinen eine heuristische Suche: Wir suchen nach einer guten Lösung, aber es ist oft nicht machbar, die optimale Lösung zu finden. Dies ist in der Regel auch bei der Suche nach Stammbäumen der Fall, wie wir in der nächsten Einheit sehen werden.
Der Vergleich von Sequenzen ist in einer grafischen Umgebung viel einfacher, daher werden wir für diesen Teil die Software MEGA verwenden. Sie sollte auf Ihrem Computer installiert sein. In MEGA können wir Sequenzdaten aus einer FASTA-Datei laden (Data > Open > Align).
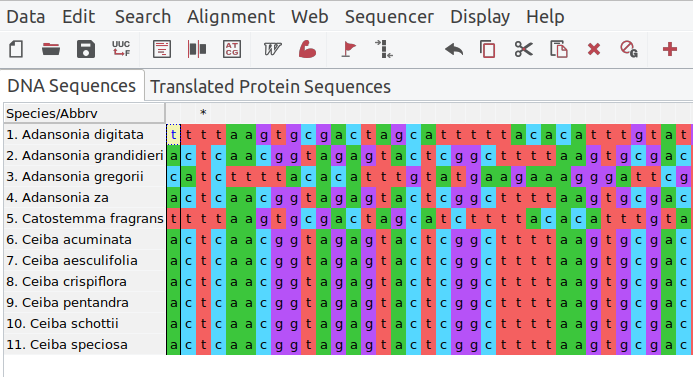
Es gibt viele verschiedene Alignment-Algorithmen. In MEGA haben wir die Wahl zwischen ClustalW und MUSCLE, die beide ähnlich gut funktionieren. Das resultierende Alignment zeigt die abgeleiteten homologen Zeichen in Spalten angeordnet, mit gaps, wo nötig. Sie werden sehen, dass einige Spalten eine große Variation aufweisen, während in anderen alle Zeichen gleich sind. Man nennt eine DNA-Position:
invariabel, wenn alle Sequenzen den gleichen Wert haben. Dies bedeutet höchstwahrscheinlich, dass die Position zwischen den Arten konserviert ist und sich nicht weiterentwickelt hat (aber können wir sicher sein?).
variabel, wenn es mindestens zwei verschiedene Proben mit unterschiedlichen Werten gibt.
Invariable Positionen helfen beim korrekten Alignment von Sequenzen, aber sie enthalten natürlich keine Informationen über Unterschiede zwischen den Arten. In MEGA können wir invariable Positionen hervorheben (Display > Toggle conserved sites > at 100% level).
Phylogenetische Software ignoriert normalerweise Lücken und mehrdeutige DNA-Zeichen oder behandelt sie als fehlende Daten (obwohl wir theoretisch das gemeinsame Vorhandensein/Fehlen von Lücken als phylogenetische Information nutzen könnten).
2.20.5.7 Variable Mutationsraten
Wir haben bereits gesehen, dass verschiedene Positionen innerhalb eines Alignments unterschiedlich variabel sind. Dies ist auf verschiedene Faktoren zurückzuführen. Ein wichtiger Faktor ist die stabilisierende Selektion, bei der Mutationen an bestimmten Stellen negative Auswirkungen haben und daher schnell verschwinden.
Die unterschiedliche Funktion verschiedener Teile von DNA-Sequenzen führt zu großen Unterschieden in der Variation:
Zwischen Genen und intergenen Regionen: Da intergene Regionen in der Regel keinen Einfluss auf den Phänotyp haben und somit nicht direkt der Selektion unterliegen, variieren sie stärker als Sequenzen in einem Gen.
Zwischen Codons innerhalb eines Gens: Sie werden sich erinnern, dass drei DNA-Basen für eine Aminosäure kodieren und ein Codon bilden. Der genetische Code ist jedoch degeneriert, was bedeutet, dass Mutationen an der dritten Stelle eines Codons in der Regel keine Auswirkungen auf das produzierte Protein haben (synonyme Mutationen). Die dritten Positionen sind daher oft sehr viel variabler als die ersten beiden.
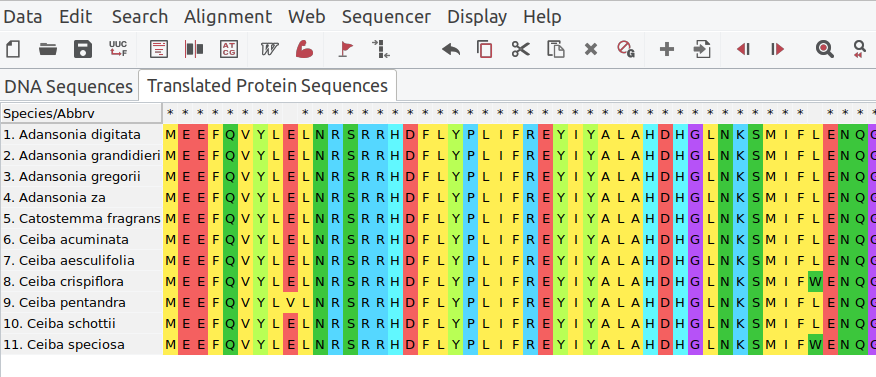
Im Beispielfall, nach Alignment der matK-Sequenzen, können wir den Beginn und das Ende des Gens finden, indem wir die Motive ATGGAGGAA (Anfang) und TCATGAATGA (Ende) suchen (Search > Motif). Diese Motive enthalten jeweils ein Startcodon (immer ATG) bzw. ein Stopcodon (mehrere möglich, hier TGA).
Wenn wir die DNA-Codons in Aminosäuren übersetzen wollen, entfernen wir zunächst die flankierenden Regionen auf der linken und rechten Seite, indem wir die Spalten auswählen und löschen. Dann können wir die entsprechenden Proteinsequenzen anzeigen lassen (Data > Translate). Da sich das Gen im Chloroplasten befindet (wir erinnern uns, dass es sich ursprünglich um ein Cyanobakterium handelte), müssen wir als genetischen Code Plant Plastid anstelle des üblichen Kern-DNA-Codes wählen.
Im Protein-Alignment erscheinen nun die Codes der 20 möglichen Aminosäuren statt der DNA-Basen:
2.20.5.8 Genetische Distanzen
Kehren wir zu R zurück. Das Paket ape hat Funktionen zum Lesen und Schreiben von DNA-Sequenzen, und wir können einige begrenzte Analysen durchführen. Eine Datei mit FASTA-Sequenzen können wir mit read.dna() einlesen. Handelt es sich dabei um ein Alignment (Sequenzen enthalten gaps und sind alle gleich lang), behandelt R das Objekt als Matrix:
library(ape)
# diese Datei enthält nur den proteinkodierenden Teil des ursprünglichen Alignments
aln <- read.dna("./data/matk_11sp_bombacoideae_cds_aligned.fas", format="fasta")
is.matrix(aln)## [1] TRUEWir können mit einer Matrix in R verschiedene Dinge anstellen, etwa uns die Anzahl der Zeilen (rows) und Spalten (columns) anzeigen lassen, oder die Zeilennamen. Wir können auch einzelne Zeilen oder Spalten auswählen oder entfernen.
## [1] 11 1515## [1] 11## [1] 1515## [1] "Adansonia_digitata" "Adansonia_grandidieri" "Adansonia_gregorii"
## [4] "Adansonia_za" "Catostemma_fragrans" "Ceiba_acuminata"
## [7] "Ceiba_aesculifolia" "Ceiba_crispiflora" "Ceiba_pentandra"
## [10] "Ceiba_schottii" "Ceiba_speciosa"## 1 DNA sequence in binary format stored in a matrix.
##
## Sequence length: 20
##
## Label:
## Adansonia_digitata
##
## Base composition:
## a c g t
## 0.40 0.05 0.25 0.30
## (Total: 20 bases)## [1] 11 50# wir entfernen alle Sequenzen, die zu Adansonia gehören
not_adansonia <- grep("Adansonia", row.names(aln), invert=TRUE)
aln_mod <- aln[not_adansonia,]
dim(aln_mod)## [1] 7 1515## [1] "Catostemma_fragrans" "Ceiba_acuminata" "Ceiba_aesculifolia"
## [4] "Ceiba_crispiflora" "Ceiba_pentandra" "Ceiba_schottii"
## [7] "Ceiba_speciosa"R komprimiert die Sequenzen standardmäßig in ein binäres Format, so dass wir die DNA-Zeichen nicht direkt sehen können. Sie können dieses Verhalten in read.dna() ändern, um die Sequenzen als Zeichenketten zu speichern (as.character = TRUE).
Wir können nun genetische Abstände zwischen unseren Sequenzen berechnen. Es gibt verschiedene Möglichkeiten, dies zu tun, wobei berücksichtigt werden kann, wie wahrscheinlich jede Art von Mutation ist. Hier berechnen wir zunächst einfach die Anzahl der Basen, die sich zwischen den einzelnen Sequenzpaaren unterscheiden:
## [1] "dist"## Adansonia_digitata Adansonia_grandidieri
## Adansonia_grandidieri 0.003963012
## Adansonia_gregorii 0.003963012 0.003963012
## Adansonia_za 0.003963012 0.002642008
## Catostemma_fragrans 0.010568032 0.010568032
## Ceiba_acuminata 0.008586526 0.008586526
## Ceiba_aesculifolia 0.009907530 0.009907530
## Ceiba_crispiflora 0.009247028 0.009247028
## Ceiba_pentandra 0.009247028 0.009247028
## Ceiba_schottii 0.009907530 0.009907530
## Ceiba_speciosa 0.008586526 0.008586526
## Adansonia_gregorii Adansonia_za Catostemma_fragrans
## Adansonia_grandidieri
## Adansonia_gregorii
## Adansonia_za 0.003963012
## Catostemma_fragrans 0.010568032 0.010568032
## Ceiba_acuminata 0.008586526 0.008586526 0.009907530
## Ceiba_aesculifolia 0.009907530 0.009907530 0.011228534
## Ceiba_crispiflora 0.007926024 0.009247028 0.010568032
## Ceiba_pentandra 0.009247028 0.009247028 0.010568032
## Ceiba_schottii 0.009907530 0.009907530 0.011228534
## Ceiba_speciosa 0.007265522 0.008586526 0.009907530
## Ceiba_acuminata Ceiba_aesculifolia Ceiba_crispiflora
## Adansonia_grandidieri
## Adansonia_gregorii
## Adansonia_za
## Catostemma_fragrans
## Ceiba_acuminata
## Ceiba_aesculifolia 0.001321004
## Ceiba_crispiflora 0.004623514 0.005944518
## Ceiba_pentandra 0.004623514 0.005944518 0.005284016
## Ceiba_schottii 0.002642008 0.003963012 0.005944518
## Ceiba_speciosa 0.003963012 0.005284016 0.000660502
## Ceiba_pentandra Ceiba_schottii
## Adansonia_grandidieri
## Adansonia_gregorii
## Adansonia_za
## Catostemma_fragrans
## Ceiba_acuminata
## Ceiba_aesculifolia
## Ceiba_crispiflora
## Ceiba_pentandra
## Ceiba_schottii 0.005944518
## Ceiba_speciosa 0.004623514 0.005284016Das Ergebnis ist eine Distanzmatrix (distance matrix oder dissimilarity matrix, in R Objektklasse dist), die paarweise Distanzen als Halbdiagonalelemente enthält.
Wir können eine solche Distanzmatrix verwenden, um ihre Elemente hierarchisch in einer baumartigen Struktur, einem Dendrogramm, zu gruppieren oder zu “clustern”. Dazu verwenden wir die R-Basisfunktion hclust mit der UPGMA-Methode, die die ähnlichsten Elemente zuerst gruppiert (also “von unten nach oben”) und davon ausgeht, dass der Abstand zwischen den Clustern die durchschnittlichen paarweisen Abstände zwischen ihren Elementen widerspiegelt.
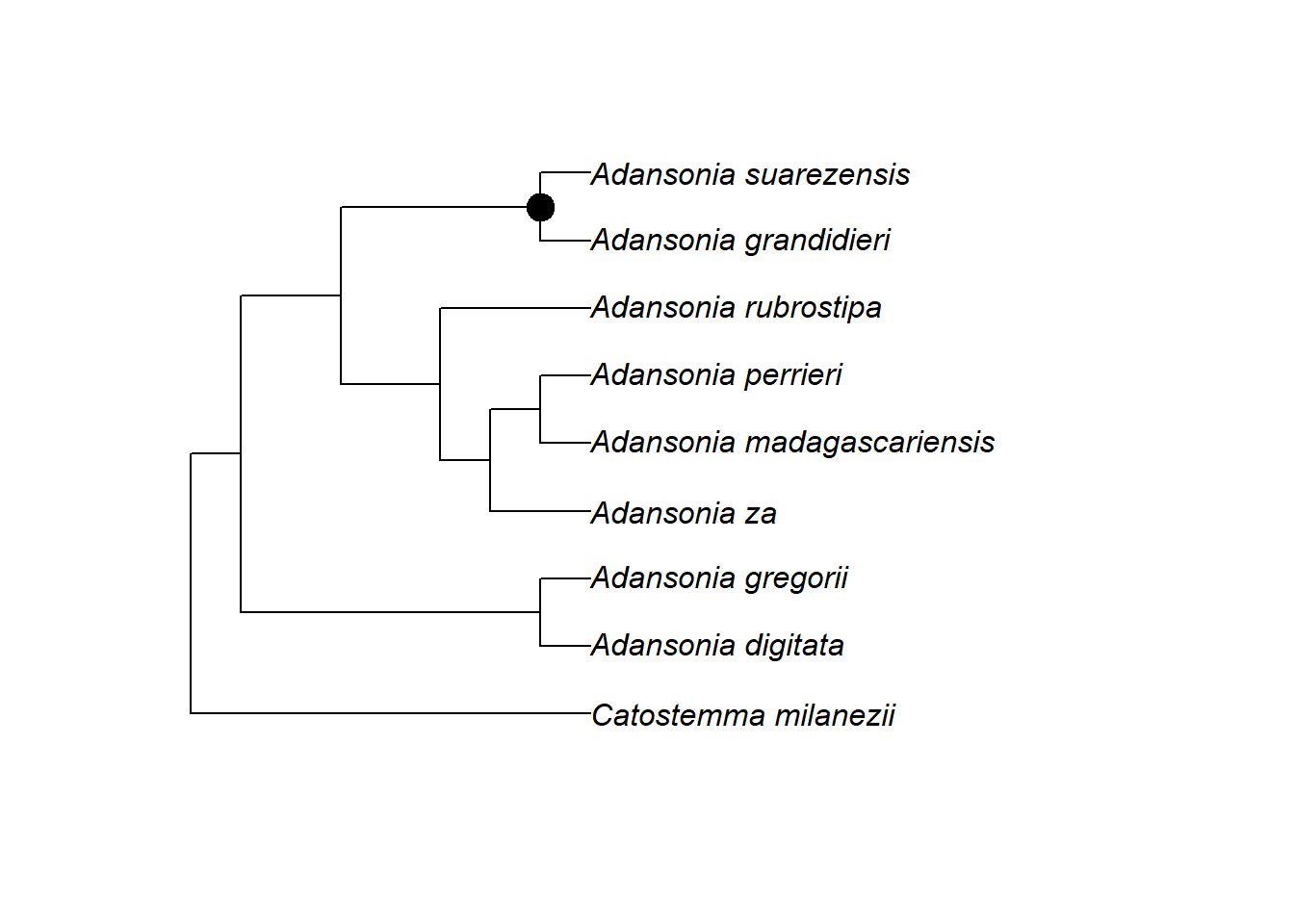
Das sieht doch einem Stammbaum schon sehr ähnlich!
Hierarchisches Clustering ist eine generische Methode, die man auf jede Art von Unähnlichkeit oder Distanz anwenden kann. In der Ökologie kann man zum Beispiel Vegetationsflächen nach der Anzahl der Arten clustern, die sich zwischen ihnen unterscheiden.
Distanzbasierte Methoden wie UPGMA sind in der Tat ein einfacher Ansatz, um aus einem DNA-Alignment einen möglichen Stammbaum abzuleiten, und sie gehörten zu den ersten verwendeten Methoden. Sie erfordern relativ wenig Berechnungen. Sie haben jedoch Mängel, und wir verwenden heute im Allgemeinen leistungsfähigere Methoden – mehr dazu in der nächsten Einheit.
2.20.6 Aufgaben
Paarweises Alignment/BLAST: In der Ergebnistabelle für die unbekannte Bombacoideae-Sequenz: Für das beste match, wieviele DNA-Basen sind unterschiedlich, und welche Art von Mutationen haben stattgefunden?
Multiples Alignment/MEGA: Erstellen Sie ein multiples Sequenzalignment der Datei matk_11sp_bombacoideae.fas mit MUSCLE. Entfernen Sie die flankierenden Regionen des Gens wie oben angegeben.
Welche Art von Mutation (Substitionen, Insertionen, Deletionen) sehen Sie innerhalb des Gens? Wie können Sie dies erklären?
An wievielen Positionen im Alignment verändert sich tatsächlich die Proteinsequenz (nicht-synonyme Mutationen)?
Was ist der Vorteil (und Nachteil) der Verwendung von proteinkodierenden Sequenzen für den Vergleich von Arten?
- Alignments in R: Lesen Sie die Datei matk_11sp_bombacoideae_cds_aligned.fas als Alignment in R ein.
- Wenn Sie von diesem Alignment nur die ersten 500 Basen verwenden, um ein UPGMA-Clustering erstellen, weicht das erhaltene Dendrogramm vom vorherigen ab? Wie lässt sich das erklären?
- Wie können wir das modifizierte Alignment nun als Datei speichern?
- Reflexion: Welche Probleme sehen Sie bei der Ableitung eines Baumes auf der Grundlage paarweiser genetischer Distanzen?